In 1906 at the University of Munich the neuropathologist Dr. Alois Alzheimer (Figure 1) was studying the brain of a recently deceased patient, Auguste Deter. Previously Alzheimer had assisted Frau Deter in the city asylum in Frankfurt and observed that she suffered from memory loss, delusions and dementia. Working in Dr. Emil Kraepelin’s lab in Munich and employing his knowledge of histopathology, Alzheimer used a silver staining technique to reveal proteinaceous senile plaques and neurofibrilliary tangles in post-mortem slices from Frau Deter’s brain.
Figure 1: Dr. Alois Alzheimer.
Shortly afterwards in the 1910 edition of the ‘Handbook of Psychiatry’ Kraepelin acknowledged his student’s contributions by naming this type of senile dementia ‘Alzheimer’s disease’ (AD).
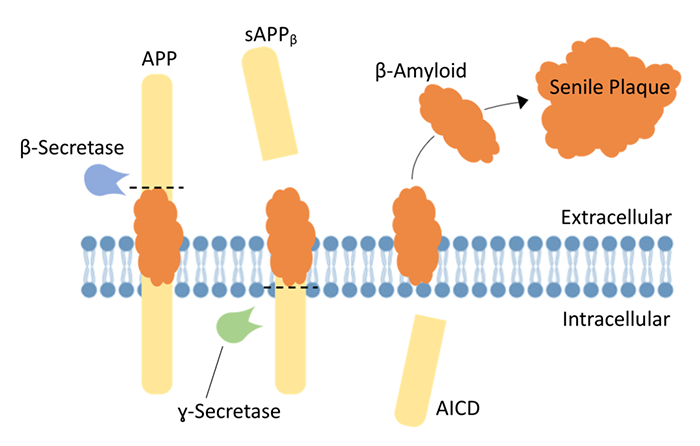
Over a century later, with an aging population and a prevalence of ~10% in people over 70, Alzheimer’s disease exerts a devastating impact on society including its sufferers, the families, healthcare systems, and the economy. However, as a result of intensive neuroscience research there have been significant advances in our understanding of the neuropathological markers first described by Alzheimer and their possible roles in disease progression. For instance, senile plaques are now recognized to be extracellular, proteinaceous aggregates predominantly composed of β-amyloid, a 40-42 amino acid peptide derived from the sequential cleaving of amyloid precursor protein (APP) by β- and ɣ-secretase enzymes (Figure 2). APP is a large membrane-bound glycoprotein that is ubiquitously expressed throughout the mammalian brain. While the role of APP in neural growth and repair has to an extent been characterized, its cleavage products, including β-amyloid, are less well understood. Low concentrations of β-amyloid production have been implicated in modulation of synaptic activity, neuroprotection and immune function within the brain. Nevertheless, when produced in excess (e.g. during AD or after traumatic brain injury) β-amyloid can promote cell damage, intracellular accumulations of neurofibrillary tangles of hyperphosphorylated tau protein, neurodegeneration and ultimately brain atrophy (Figure 3).
Figure 2: Schematic showing the processing of Amyloid Precursor protein (APP) by secretase enzymes to generate β-amyloid. β-secretase cleaves APP to release soluble APPβ (sAPPβ) extracellularly. The residual transmembrane protein is then subsequently cleaved by ɣ-secretase to release APP intracellular domain (AICD) and toxic forms of β-amyloid extracellularly. In excess the β-amyloid is sequestered into aggregates that Dr. Alzheimer observed and described as ‘senile plaques.’
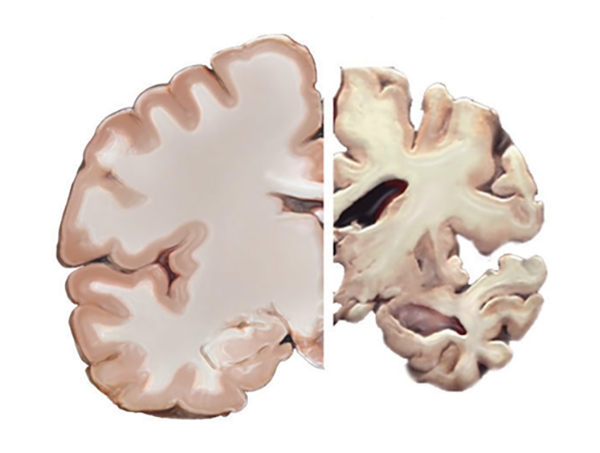
Figure 3: Gross atrophy from Alzheimer’s disease. On the left, a healthy coronal brain section. On the right, a hemisphere ravaged through AD-related neurodegeneration.
Based on our current knowledge of the molecular pathways leading to pathology and cognitive decline, researchers have developed transgenic mouse models of AD that provide the neuroscience community important tools to study disease progression and to explore potential therapies. Strategies for developing such models have included introducing mutated genes into the mouse brain that are responsible for familial forms of early onset Alzheimer’s disease in humans. For instance, the 5xFAD mouse (Oakley et al., J. Neurosci, 2006) harbors multiple mutations in genes encoding for human APP and Presenilin-1, a key component of the ɣ-secretase enzyme complex. The expression of the mutant APP and the aberrant processing by mutant ɣ-secretase result in excessive production of neurotoxic levels of β-amyloid 42. In turn this leads to extensive cerebral amyloidosis (deposition of senile plaques) and pronounced gliosis, whereby reactive astrocytes migrate to damaged brain regions. Thus, the 5xFAD mouse recapitulates many AD-phenotypes including widespread neurodegeneration and cognitive deficits as early as 6 months postnatally.
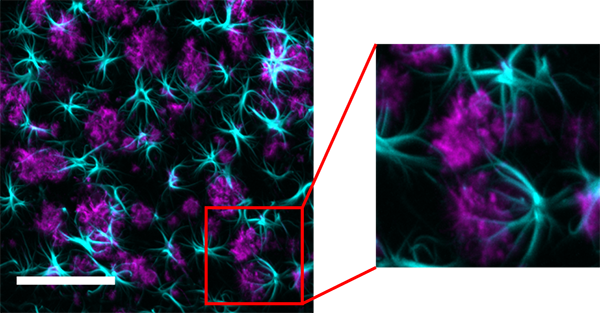
At LifeCanvas Technologies using rapid clearing through our SmartClear II Pro device and uniform immunolabeling with our SmartLabel device, we processed samples prepared from the intact brains of 5xFAD mice from Dr. Jonathan Epp’s lab in the Department of Cell Biology and Anatomy, University of Calgary. Dr. Epp and his lab are primarily interested in the neural mechanisms that underlie memory consolidation and the pathological processes that interrupt memory processing. Dr. Epp’s 5xFAD mouse brain samples were immunolabeled for β-amyloid and for GFAP (to tag astrocytes) and 3D image data were acquired using a 15X objective (Applied Scientific Instrumentation) and our SmartSPIM lightsheet microscope (Figure 4). At this resolution, astrocytes are clearly observed to be extending their processes towards the amyloid aggregates in order to clear and degrade these deposits. No doubt Alzheimer would have relished these spectacular, high resolution images of reactive astrocytes surrounding senile plaques that lend a 3D perspective on the pathological markers that he had originally revealed and described.
Figure 4: GFAP (cyan) and β-amyloid (magenta) of a 5xFAD mouse brain labeled and imaged at LifeCanvas Technologies using our SmartClear II Pro, SmartLabel, and SmartSPIM devices, with a 15X objective (ASI). Scale bar: 100 µm. Tissue courtesy of the Hotchkiss Brain Institute – Advanced Microscopy Platform and the Lab of Dr. Jonathan Epp, Dept. of Cell Biology & Anatomy, University of Calgary.