

LifeCanvas (LC): What is your research background, and what led you to the Joslin Diabetes Center?
Wenqiang Chen (WC): In 2017, I started my first post-doc position in Dr. Yi Zhang’s lab at Boston Children’s Hospital and Harvard Medical School, where I worked on mouse models of drug addiction. We used tools like single-cell sequencing and in vivo calcium imaging to look at potential contributions of different brain clusters to cocaine addiction.
I have always been interested in disease research, but I wanted an opportunity to collaborate with researchers directly facing clinics. Now, I am working in Dr. C. Ronald Kahn’s lab at the Joslin Diabetes Center at Harvard Medical School, where I have opportunities to access many resources for human samples, clinical data, and patient-derived cell lines. Many people in the lab are working on patient-derived iPSCs, differentiating them into various cell types to study diabetes-in-a-dish.

LC: Your recent publication examines the interaction of Type II Diabetes and Alzheimer's. How did you become interested in this overlap?
There is compelling clinical evidence suggesting a link between insulin resistance and Alzheimer’s disease – some people are even starting to call Alzheimer’s disease “Type 3 Diabetes”. We know that insulin resistance may hasten the development or increase the risk of Alzheimer’s, and that people with Alzheimer’s are more likely to develop insulin resistance. Studies have also shown that diabetes can worsen aging-related memory loss, and there are many other features shared by both conditions, including: oxidative stress, mitochondrial dysfunction, disruption of the blood-brain barrier, and inflammation. So, there must be molecular links between these two chronic conditions.
Clinical trials have shown that intranasal insulin delivery can reduce memory dysfunction in Alzheimer’s patients, as it can bypass the blood-brain-barrier. Animal studies also suggest that counteracting cell-type-specific effects of insulin resistance can reduce brain inflammation and other characteristics of Alzheimer’s. In my Trends in Neurosciences review paper, I summarize some of these interesting clinical trials at the intersection of insulin resistance and neurodegeneration.
LC: How did you choose to focus on astrocytes?
WC: Studies have identified disease-associated astrocytes and microglia, which have distinct transcriptional features from other cells of the same type. Over twenty years ago, our lab developed a whole-brain insulin receptor knockout with Dr. Jens Bruning, who is now directing the Max-Planck institute of Metabolism Research. In a 2018 study, the Kahn lab also developed a mouse model with an astrocyte-specific insulin receptor knockout. We wanted to combine both of these knockouts in the 5xFAD Alzheimer’s mouse model to see whether we could induce a stronger Alzheimer’s phenotype.
LC: What are the most significant changes you saw in insulin-resistant Alzheimer's model mice?
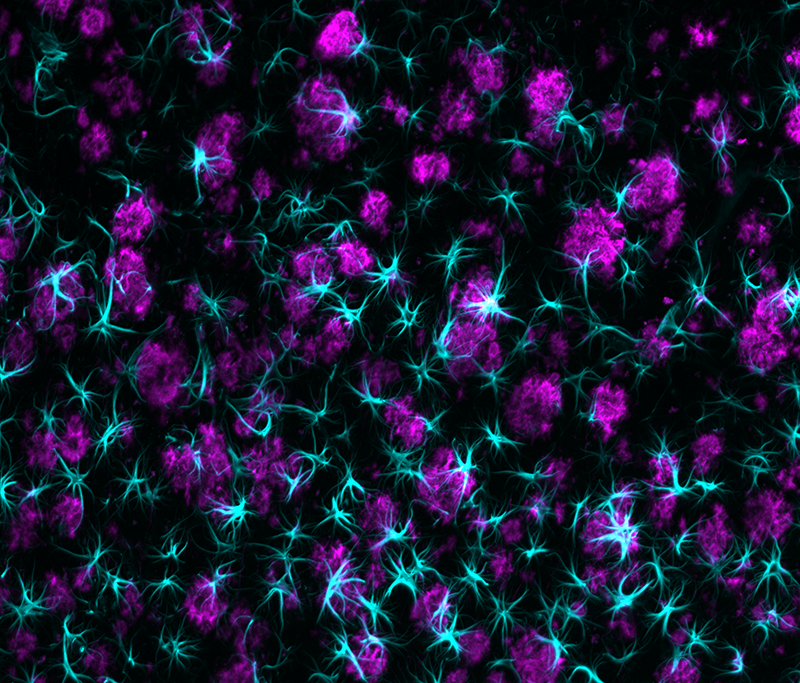
WC: We consistently see that loss of insulin signaling in astrocytes can induce impairments in the uptake of beta-amyloid plaques, which are associated with Alzheimer’s disease. We assessed the levels of fluorescently-tagged beta-amyloid peptides with flow cytometry, and also used whole-brain mapping to look at the extent of astrocytes surrounding the beta-amyloid aggregates. All of our methods show that the impairment of insulin signaling in astrocytes can significantly inhibit beta-amyloid uptake.
LC: Do you plan to also study the contributions of microglia to these combined disease phenotypes?
WC: My current project focuses on microglia. Many studies have shown that microglia may play a more important role in the progression of Alzheimer’s disease than astrocytes. Thus microglia knockouts may show a much stronger phenotype than astrocyte knockouts, which could further contribute to our understanding of the interaction between type II diabetes and Alzheimer’s disease. I have already sent samples from microglia knockout 5xFAD mice to LifeCanvas to better assess this hypothesis.
LC: The paper suggests a potential feedback mechanism between beta-amyloid and insulin signaling in astrocytes. Could you elaborate?
WC: Our results show that insulin signaling can be disrupted by the presence of beta-amyloid plaques. We assessed this by using Western blots to look at the phosphorylation levels of proteins involved in insulin signaling pathways. We incubated isolated, regular astrocytes (no impairment in insulin signaling) with beta-amyloid, and with or without insulin stimulation. Usually, in the presence of insulin, we can see clear phosphorylation of insulin signaling proteins. But in the presence of beta-amyloid, some of the proteins show consistently lower levels of phosphorylation. This suggests that beta-amyloid peptides can interact with astrocytes or other mechanisms to disrupt insulin signaling.
LC: What role have LifeCanvas’ whole-brain mapping tools played in your research?
WC: Initially, we used more traditional approaches – namely immunohistochemistry (IHC) and Western blots – to look at the distribution levels of soluble and insoluble beta-amyloid protein deposits. Western blots require ultracentrifugation of entire brain hemispheres, which gives you total beta-amyloid levels per hemisphere, but completely erases region-specific nuances. IHC can reveal region-specific information, but it’s also very tedious, time-consuming, and low-throughput: you need to section samples, and then stain all of them.
"LifeCanvas’ unbiased screening approach ... provides a more holistic picture of how different brain regions may contribute to the spread of beta-amyloid deposition."
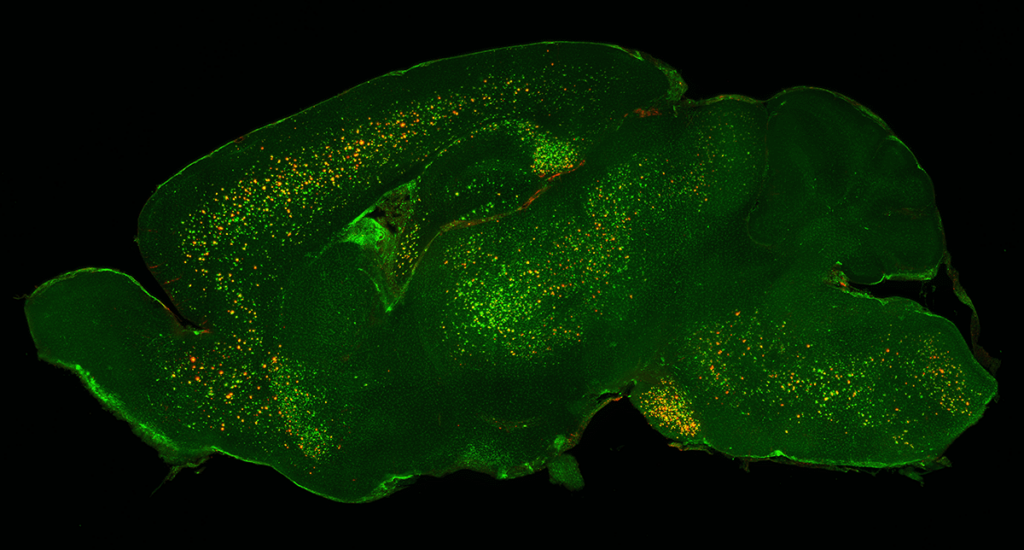
I first met LifeCanvas scientists in 2019 at the Society for Neuroscience conference in Chicago, and decided to pursue a pilot study using 3D whole-brain mapping technology. In addition to providing a high-throughput pipeline for obtaining regional data throughout the entire brain, this technology also offers an unbiased look at the spatial distribution of beta-amyloid and astrocytes.
Many studies in Alzheimer’s disease are driven by a preexisting hypothesis of which regions to focus on, such as the hippocampus or the prefrontal cortex. Personally, I prefer LifeCanvas’ unbiased screening approach, because it provides a more holistic picture of how different brain regions may contribute to the spread of beta-amyloid deposition. Sometimes we can discover unexpected results through this approach.
LC: What brain regions did you focus on, and what are you excited to explore next?
WC: In this paper, we looked at more conventional regions like the cortex, which is large and has a strong beta-amyloid deposition phenotype. However, in my ongoing microglia study, I used LifeCanvas-generated heat maps to discover more interesting regions, including the basolateral amygdala and the subiculum. We actually saw that the amygdala showed the most specific microglia activation pattern when we knocked out insulin receptors, suggesting it may be more vulnerable to insulin disruption.
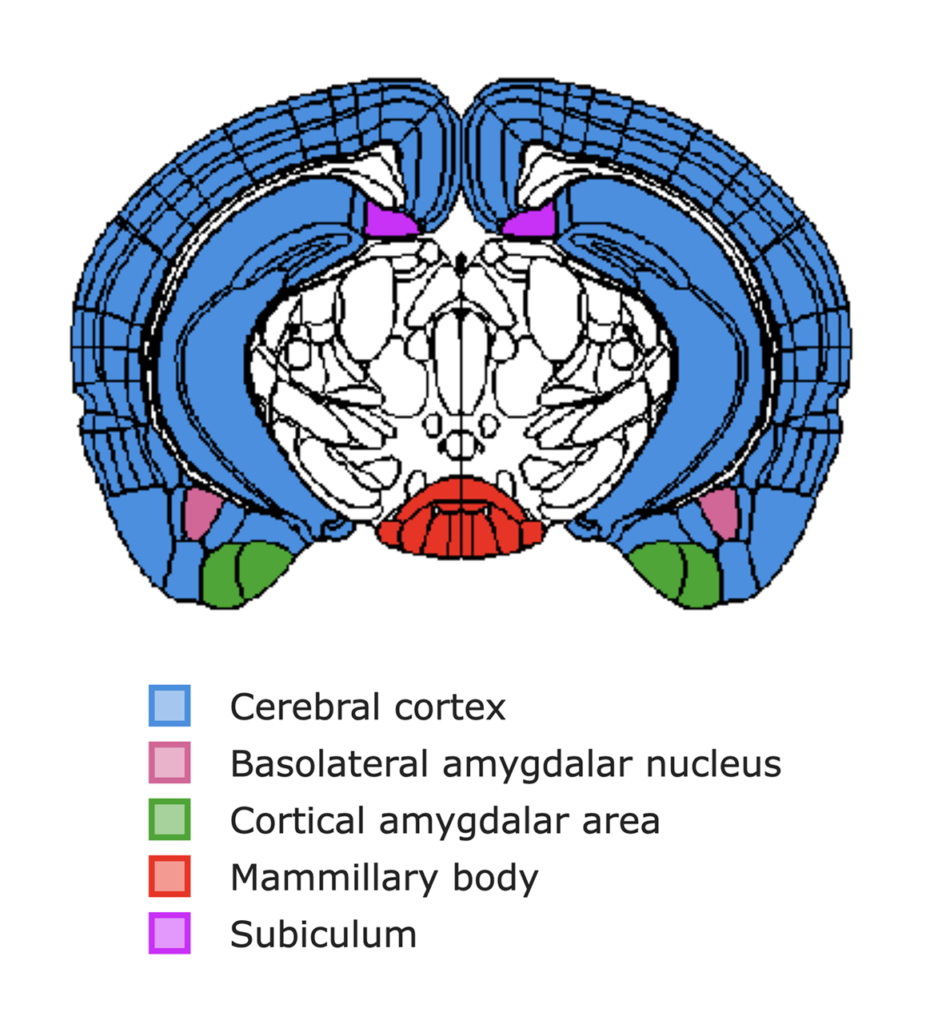
We also saw a lot of microglia activation in the subiculum in the hippocampal formation, which other labs have suggested as a potential inception point for beta-amyloid deposition. Additionally, Dr. Li-Huei Tsai’s lab at MIT has shown that a region called the mammillary body may be one of the earliest sites of neurodegeneration in Alzheimer’s disease.
Another interesting result I gleaned from the whole-brain data is that some regions actually appear to be free of beta-amyloid deposition, while still showing highly specific microglia activation patterns. This is still an ongoing investigation.
In the future, I want to look at how microglia contribute to phagocytosis (or clearance) of beta-amyloid, and how this is affected by insulin resistance. We plan to explore this by examining several factors: how microglia move towards and surround plaques in 3D, the percentage of engulfment of plaques, and the presence of neural death markers coincident with microglia that can indicate problems in phagocytosis. This is a fascinating area that I would like to pursue as a life-long career.
