
Antibody Validation
For this protocol, you will need at least three 100-200 µm thick tissue sections. We recommend obtaining these sections with a vibratome or with our product, Megatome. You will need at least one slice that is only PFA fixed, and at least two slices that were SHIELD fixed and delipidated. You can take multiple slices from a PFA fixed brain, and SHIELD and Delipidate some of them using the thin slice protocol. Alternatively, you can take some slices from a PFA fixed brain, and some slices from a brain that was SHIELD fixed and delipidated using the standard protocols.
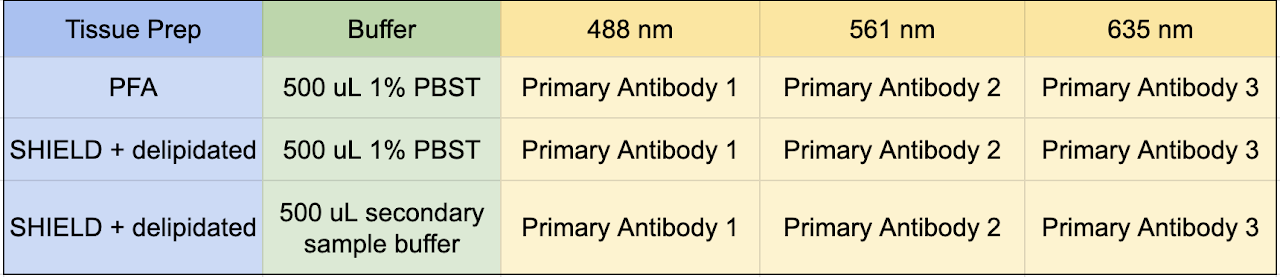
This protocol allows you to test three antibodies. The matrix below allows you to control for SHIELD fixing and delipidating your tissues, as well as staining with our sample buffers as opposed to 1% PBST.
[Note] This protocol will give a good idea of the compatibility of the antibody, but it will not guarantee success in whole tissue or in the active labeling condition with the electric field. After successfully completing this protocol, you can move to test a whole sample with electrophoretic labeling.
Reagents
- PBST (PBS with 1% TritonX-100)
- Secondary Sample Buffer
- PFA Fixed brains
- Well Plate
- Well Plate cover
Protocol
PASSIVE LABELING
1. Wash all tissue sections thoroughly in 1% PBST.
- Use a well plate to do 3 washes at room temperature, a minimum 10 minutes per wash.
2. Fill a set of new wells with 500 µl of 1% PBST or 500 µl of Secondary Sample Buffer as needed.
3. Add primary antibodies and dyes to each well according to the manufacturer’s recommended dilution.
4. Using a paint brush, carefully move each tissue slice over into the new wells.
5. Place sealing tape over the well plate and close it. Wrap the plate in aluminum foil or place in a black sample bag to protect it from light.
6. Shake gently at room temperature overnight.
7. The following day, remove the buffer and unbound antibodies using a pipette and repeat the washing steps described above (3x 10 minutes). Make sure to use the same buffers for the washes that the tissue sections have been incubating in during the labeling step (1% PBST or Secondary Sample buffer). Keep the plate protected from light.
Add fresh PBST or Secondary Sample Buffer to a new set of wells (500 µL per well) as needed and add secondary antibodies again at the recommended dilution.
Replace the sealing tape and shake the well plate at room temperature for several hours or overnight.
- Repeat the washing steps described above to wash away unbound secondary antibody. Image immediately or move sections to centrifuge tubes with PBST at room temperature, protected from light, to store for a short time. Antibodies can dissociate over time, so it is best to image within a day of labeling.
- Note: You may combine primary and secondary antibodies in a single labeling step to save time. However, we tend to get better results with sequential labeling, and it is more comparable to the SmartLabel/SmartBatch+ sequential labeling protocol for whole samples.
Interpretation of Results
There are a few things you should look out for while imaging:
- If you see strong positive signal in all samples, it is likely that your antibody is compatible with SHIELD, delipidation, and labeling buffers. The next step in this case is to try an active labeling experiment to confirm and evaluate for labeling uniformity and brightness.
- If there is no positive signal in the control sample, then the antibody may not be compatible with your tissue type.
- [Tip] Alternatively, your antibody may be expired.
- If the signal is worse in the SHIELD + delipidated sample compared to the control it is possible that the SHIELD fixation could be blocking the binding site, or that the delipidation step could be damaging the epitope, although this is less likely.
- If the signal is worse (dimmer signal, or excess nonspecific signal) in Secondary Sample Buffer compared to PBST, then it may not be compatible with the buffers.